3.1. Simulation of the oxidation of methane
Jinzhe Zeng, Liqun Cao, and Tong Zhu
This tutorial was adapted from: Jinzhe Zeng, Liqun Cao, Tong Zhu (2022), Neural network potentials, Pavlo O. Dral (Eds.), Quantum Chemistry in the Age of Machine Learning, Elsevier. Please cite the above chapter if you follow the tutorial.
In this tutorial, we will take the simulation of methane combustion as an example and introduce the procedure of DP-based MD simulation. All files needed in this section can be downloaded from tongzhugroup/Chapter13-tutorial. Besides DeePMD-kit (with LAMMPS), ReacNetGenerator should also be installed.
3.1.1. Step 1: Preparing the reference dataset
In the reference dataset preparation process, one also has to consider the expect accuracy of the final model, or at what QM level one should label the data. In this paper, the Gaussian software was used to calculate the potential energy and atomic forces of the reference data at the MN15/6-31G** level. The MN15 functional was employed because it has good accuracy for both multi-reference and single-reference systems, which is essential for our system as we have to deal with a lot of radicals and their reactions. Here we assume that the dataset is prepared in advance, which can be downloaded from tongzhugroup/Chapter13-tutorial.
3.1.2. Step 2. Training the Deep Potential (DP)
Before the training process, we need to prepare an input file called methane_param.json which contains the control parameters. The training can be done by the following command:
$ $deepmd_root/bin/dp train methane_param.json
There are several parameters we need to define in the methane_param.json file. The type_map refers to the type of elements included in the training, and the option of rcut is the cut-off radius which controls the description of the environment around the center atom. The type of descriptor is se_a in this example, which represents the DeepPot-SE model. The descriptor will decay smoothly from rcut_smth (R_on) to the cut-off radius rcut (R_off). Here rcut_smth and rcut are set to 1.0 Å and 6.0 Å respectively. The sel defines the maximum possible number of neighbors for the corresponding element within the cut-off radius. The options neuron in descriptor and fitting_net is used to determine the shape of the embedding neural network and the fitting network, which are set to (25, 50, 100) and (240, 240, 240) respectively. The value of axis_neuron represents the size of the embedding matrix, which was set to 12.
3.1.3. Step 3: Freeze the model
This step is to extract the trained neural network model. To freeze the model, the following command will be executed:
$ $deepmd_root/bin/dp freeze -o graph.pb
A file called graph.pb can be found in the training folder. Then the frozen model can be compressed:
$ $deepmd_root/bin/dp compress -i graph.pb -o graph_compressed.pb -t methane_param.json
3.1.4. Step 4: Running MD simulation based on the DP
The frozen model can be used to run reactive MD simulations to explore the detailed reaction mechanism of methane combustion. The MD engine is provided by the LAMMPS software. Here we use the same system from our previous work, which contains 100 methane and 200 oxygen molecules. The MD will be performed under the NVT ensemble at 3000 K for 1 ns. The LAMMPS program can be invoked by the following command:
$ $deepmd_root/bin/lmp -i input.lammps
The input.lammps is the input file that controls the MD simulation in detail, technique details can be found in the manual of LAMMPS. To use the DP, the pair_style option in this input should be specified as follows:
pair_style deepmd graph_compressed.pb
pair_coeff * *
3.1.5. Step 5: Analysis of the trajectory
After the simulation is done, we can use the ReacNetGenerator software which was developed in our previous study to extract the reaction network from the trajectory. All species and reactions in the trajectory will be put on an interactive web page where we can analyze them by mouse clicks. Eventually we should be able to obtain reaction networks that consistent with the following figure.
$ reacnetgenerator -i methane.lammpstrj -a C H O --dump
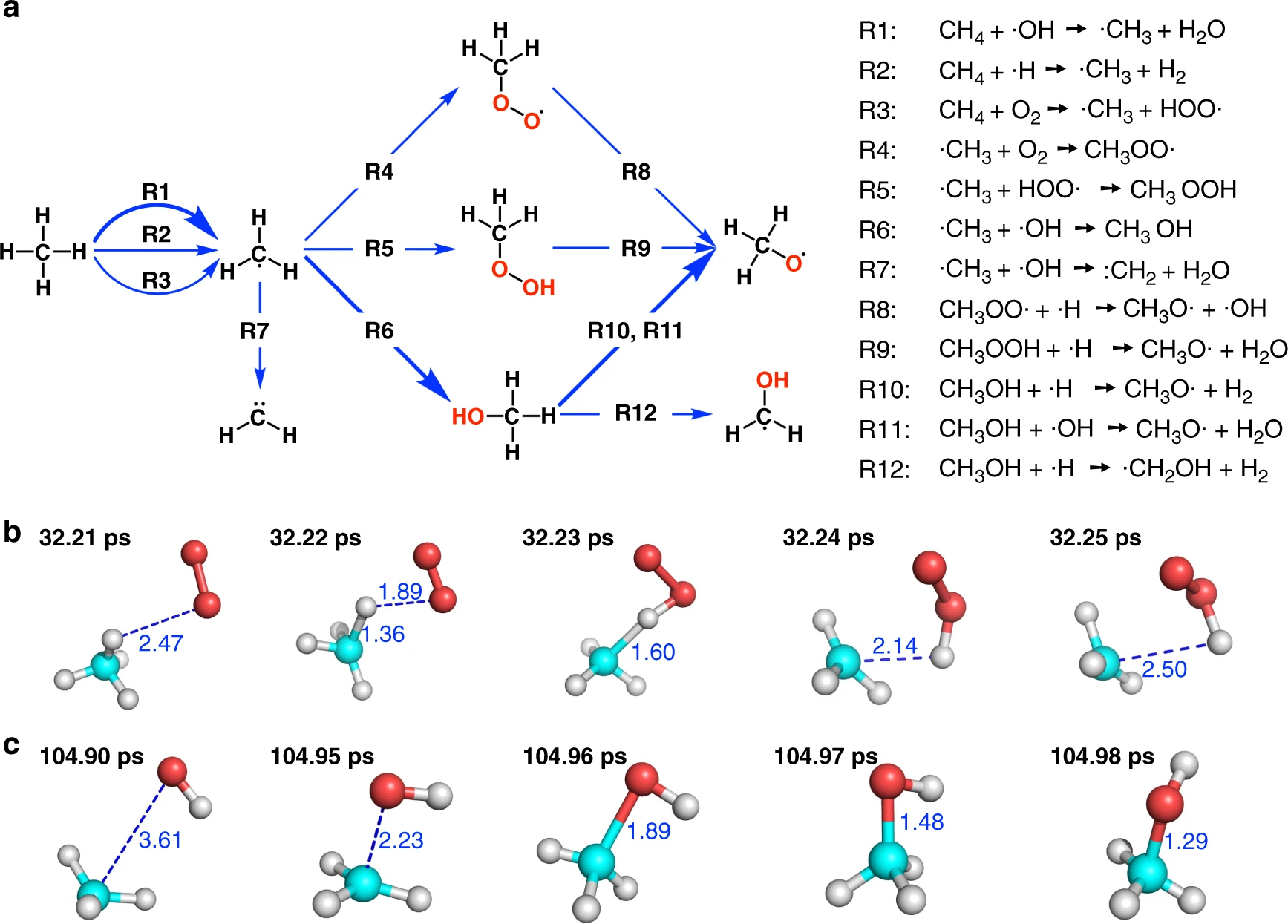
Fig: The initial stage of combustion. The figure is taken from this paper and more results can be found there.
3.1.6. Acknowledge
This work was supported by the National Natural Science Foundation of China (Grants No. 22173032, 21933010). J.Z. was supported in part by the National Institutes of Health (GM107485) under the direction of Darrin M. York. We also thank the ECNU Multifunctional Platform for Innovation (No. 001) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant ACI-1548562.56 (specifically, the resources EXPANSE at SDSC through allocation TG-CHE190067), for providing supercomputer time.
3.1.7. References
Jinzhe Zeng, Liqun Cao, Tong Zhu (2022), Neural network potentials, Pavlo O. Dral (Eds.), Quantum Chemistry in the Age of Machine Learning, Elsevier.
Jinzhe Zeng, Liqun Cao, Mingyuan Xu, Tong Zhu, John Z. H. Zhang, Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation, Nature Communications, 2020, 11, 5713.
Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H., Gaussian 16, revision A. 03. Gaussian Inc., Wallingford CT 2016.
Han Wang, Linfeng Zhang, Jiequn Han, Weinan E, DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics, Computer Physics Communications, 2018, 228, 178-184.
Aidan P. Thompson, H. Metin Aktulga, Richard Berger, Dan S. Bolintineanu, W. Michael Brown, Paul S. Crozier, Pieter J. in ‘t Veld, Axel Kohlmeyer, Stan G. Moore, Trung Dac Nguyen, Ray Shan, Mark J. Stevens, Julien Tranchida, Christian Trott, Steven J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Communications, 2022, 271, 108171.
Denghui Lu, Wanrun Jiang, Yixiao Chen, Linfeng Zhang, Weile Jia, Han Wang, Mohan Chen, DP Train, then DP Compress: Model Compression in Deep Potential Molecular Dynamics, 2021.
Jinzhe Zeng, Liqun Cao, Chih-Hao Chin, Haisheng Ren, John Z. H. Zhang, Tong Zhu, ReacNetGenerator: an automatic reaction network generator for reactive molecular dynamics simulations, Phys. Chem. Chem. Phys., 2020, 22 (2), 683–691.